В дефицит на α-N-ацетилгалактозаминидаза това е заболяване на лизозомното съхранение, което се среща много рядко. Разделя се на юношеска и възрастна форма.
Какво е дефицит на a-N-ацетилгалактозаминидаза?

Дефицитът на a-N-ацетилгалактозаминидаза или Дефицит на алфа-N-ацетилгалактозаминидаза се нарича още в медицината Болест на Шиндлер, Болест на Шиндлер или Болест на Канзаки обозначен. Има предвид изключително рядко заболяване на лизозомно съхранение, наследяването на което е автозомно рецесивно. Лекарите разграничават юношеската форма, която се среща при деца и юноши, и възрастната форма, от която страдат възрастните.
Юношеската форма на дефицит на алфа-N-ацетилгалактозаминамидаза се нарича болест на Шиндлер, докато възрастната форма се нарича болест на Канзаки. Но има и подразделения на болестта на Шиндлер тип I за младежката форма и болестта на Шиндлер тип II за възрастната форма. Възможна е и смес от двете форми, известна като болест на Шиндлер тип III.
И трите форми на заболяването са олигозахаридози (гликопротеинози), които включват фукозидоза и съхранение на сиалова киселина. Името на болестта на Шиндлер или болестта на Шиндлер за дефицит на алфа-N-ацетилгалактозаминидаза върви към германския генетик Детлев Шиндлер.
Той е първият, който описва заболяването през 1988 г. Година по-късно японският лекар Т. Канзаки описва формата на възрастните. Той открива през 1991 г., че дефицитът на алфа-N-ацетилгалактозаминамидаза е причината за заболяването.
каузи
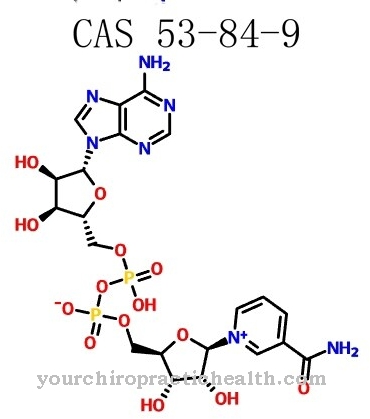
Дефицитът на α-N-ацетилгалактозаминидаза се причинява от намалена активност на ензима алфа-N-ацетилгалактозаминидаза. Мутантни или глупостни мутации на гена NAGA, който кодира алфа-N-ацетилгалактозаминамидаза, са отговорни за ензимния дефект.
NAGA генът се намира на хромозома 22 ген локус q11. Ензимът алфа-N-ацетилгалактозаминидаза има за задача да катализира разцепването на N-ацетилгалактозамина от различни гликолипиди и гликопротеини. Генетичният дефект причинява намалена активност на ензима, така че вещества, които не се метаболизират, се натрупват в клетките на тялото.
В повечето случаи това са протеогликани, гликосфинголипиди и N- или О-свързани гликопротеини. Натрупването на тези причини причинява увреждане на клетката и засегнатите органи. Недостигът на алфа-N-ацетилгалактозаминидаза е толкова рядък, че няма точна информация за честотата му.
Досега са известни само дванадесет пациенти от осем семейства, които са развили болестта на Шиндлер в света. Неврологичните оплаквания не се срещат при всеки пациент. Някои лекари подозират влиянието на допълнителни фактори за появата на неврологични симптоми. Все още обаче няма доказателства в подкрепа на това предположение.
Можете да намерите лекарствата си тук
➔ Лекарства за болкаСимптоми, заболявания и признаци
Симптомите на дефицит на алфа-N-ацетилгалактозаминидаза зависят от конкретната форма на заболяването. В контекста на болестта на Шиндлер тип I, прогресиращата мускулна хипотония се появява през първата година от живота. Освен това засегнатите бебета страдат от изразена психомоторна регресия.
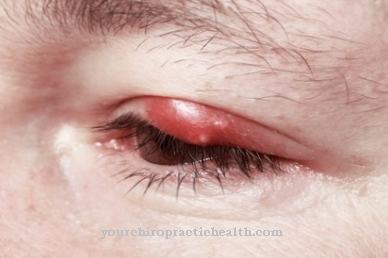
Нарушения в последователността на движение, спастична тетраплегия и миоклонични мозъчни припадъци също са регистрирани като симптоми. Съществува и риск детето да ослепее. Ако има възрастна форма на дефицит на алфа-N-ацетилгалактозаминамидаза, т.е. болест на Шиндлер тип 2, се появяват симптоми, подобни на болестта на Фабри.
Пациентите имат ангиокератоми, които са доброкачествени кожни промени. Много пациенти са леко умствено изостанали. Клинична междинна форма е болест на Шиндлер тип III. Засегнатите страдат от неврологични неизправности, мозъчни припадъци и умствени увреждания.
Възможни са и по-малко тежки форми, които са свързани с леки психиатрични и неврологични оплаквания, забавено езиково развитие или симптоми, подобни на аутизъм.
Диагноза и курс
Диагнозата на дефицит на алфа-N-ацетилгалактикозаминидаза е възможна чрез откриване на намалена активност на NAGA. За тази цел се провеждат ензимни тестове в кръвна плазма, левкоцити или култивирани фибробласти или лимфобласти. Хроматографското откриване на олигозахариди в урината също дава възможност за диагностициране на ензимния дефицит.
Може да се направи и ДНК анализ, но това не е необходимо в повечето случаи. По време на бременността пренаталната диагноза на ензимния дефицит може да се извърши и с помощта на мутационен анализ на гена NAGA след вземане на проба от хорионни въшки или амниоцентеза. Съответната форма на заболяването обаче не може да бъде определена.
Диференциалната диагноза на синдрома на Фабри, свързаната с пантотенат киназа невродегенерация и инфантилна невроаксонална дистрофия, които също имат ензимни дефекти, също са важни. Ходът на дефицит на алфа-N-ацетилгалактозаминамидаза зависи от съответната форма на заболяването.
Заболяването на Шиндлер тип I се счита за неблагоприятно, докато прогнозата за другите два вида е по-благоприятна. Броят на болни хора обаче е толкова малък, че не могат да се правят смислени твърдения относно продължителността на живота.
Усложнения
Симптомите и усложненията на дефицита на A-N-ацетилгалактозаминамидаза силно зависят от неговата форма.В повечето случаи обаче се наблюдават нарушения в движението, така че засегнатите са относително ограничени в ежедневието си. Появяват се и спазми, които са свързани със силна болка.
Бебетата по-специално страдат от тежки нарушения в развитието, които могат да доведат до спастичност при детето. В някои случаи дефицитът на A-N-ацетилгалактозаминамидаза води до пълна слепота при пациента. Регресията на интелигентността води до забавяне и по този начин до увреждания.
Те обикновено ограничават значително ежедневието на пациента и имат отрицателен ефект върху качеството на живот. Често засегнатите са зависими от помощта на други хора. Забавянето може също да доведе до разстройство на намирането на думи или нарушение на езика. Не е възможно да се лекува дефицитът на A-N-ацетилгалактозаминамидаза причинно.
Поради тази причина засегнатото лице трябва да приема антибиотици. Някои усложнения също трябва да се избягват директно. Пневмонията например е една от тях. Освен това пациентът може да зависи и от изкуственото хранене.
Кога трябва да отидете на лекар?
Дефицитът на A-N-ацетилгалактозаминидаза определено трябва да се лекува от лекар. Този дефицит не отшумява сам по себе си и няма спонтанно изцеление на това заболяване. Във всеки случай трябва да се консултирате с лекар, ако родителите забележат спад на двигателните и умствените способности при своето дете или бебе. Оплакванията с прости движения или процеси също могат да показват дефицит на A-N-ацетилгалактозаминидаза и трябва да бъдат прегледани от лекар.
Освен това различни спастичности също са признак на заболяване. Не са рядкост пациентите да страдат от интелектуални затруднения и други неизправности. Ако тези оплаквания са налице, определено е необходимо лечение. Ако няма лечение, това може да доведе до значителни оплаквания и усложнения в зряла възраст и по този начин значително да намали качеството на живот на засегнатото лице.
Силно забавеното развитие на речта също може да е признак на дефицит на A-N-ацетилгалактозаминидаза и затова трябва да бъде изследвано. Като правило може да се консултира с общопрактикуващ лекар, за да се определи дефицитът на A-N-ацетилгалактозаминамидаза. След това по-нататъшното лечение на отделните оплаквания се извършва от специалист.
Лекари и терапевти във вашия район
Лечение и терапия
Причинно-следствената терапия за дефицит на алфа-N-ацетилгалактозаминидаза не е възможна. Следователно, медицинският подход е ограничен до лечение на симптомите. Това включва разумна диета и прием на течности, предотвратяване на инфекциозни заболявания, което може да се извърши и чрез прилагане на антибиотици, и ограничаване на церебрални атаки чрез прилагане на антиепилептични лекарства.
По-нататъшните стъпки на лечение включват физиотерапевтични мерки за предотвратяване на пневмония и контрактури и за намаляване на болката или спастичността с помощта на лекарства. Освен това аспирационната профилактика може да се проведе чрез изкуствено хранене.
Последните проучвания показват, че дефицитът на α-N-ацетилгалактозаминамидаза е нарушение на сгъването на протеини, така че ензимната заместителна терапия се обсъжда като разумна мярка за лечение. Генната терапия е друг възможен терапевтичен подход за лечение на нарушения на лизозомното съхранение. Тъй като болестта на Шиндлер се наследява по автозомно рецесивен начин, се препоръчва генетично консултиране на засегнатите семейства.
Прогноза и прогноза
Дефицитът на A-N-ацетилгалактозаминидаза може да доведе до различни симптоми. В повечето случаи обаче дефицитът води до забавено психомоторно развитие. В зряла възраст пациентът често е зависим от помощта на други хора и не може сам да се справи с ежедневието. Появяват се и нарушения в движението. Това може да се забележи чрез накуцване или нарушения на координацията.
В най-лошия случай пациентът може да ослепее или да страда от тежки зрителни нарушения поради дефицита на A-N-ацетилгалактозаминамидаза. Не са редки случаите на интелектуални увреждания, които значително намаляват качеството на живот на съответния човек. Появяват се и нарушения на речта и разстройство на намирането на думи, които могат да повлияят на живота. По-специално децата са засегнати от тормоз и дразнене поради симптомите на дефицит на A-N-ацетилгалактозаминамидаза.
Причинно-следственото лечение на дефицита на A-N-ацетилгалактозаминамидаза не е възможно, така че лечението е насочено основно към намаляване на симптомите. За намаляване на нарушенията в развитието могат да се използват различни терапевтични мерки. В много случаи обаче пациентът е зависим от външна помощ в ежедневието. Продължителността на живота не се влияе от дефицита.
Можете да намерите лекарствата си тук
➔ Лекарства за болкапредотвратяване
Не са известни превантивни мерки срещу дефицит на алфа-N-ацетилгалактозаминамидаза. Болестта на Шиндлер е едно от изключително редките заболявания.
Aftercare
Тъй като дефицитът на A-N-ацетилгалактозаминидаза е вродено заболяване, което не може да се лекува причинно, а само симптоматично, цялостно лечение не е възможно. Засегнатият човек е зависим от лечението през целия живот, поради което възможностите за последваща грижа са много ограничени.
По правило поради дефицита на A-N-ацетилгалактозаминамидаза пациентът често зависи от приема на лекарства и антибиотици. Възможните взаимодействия с други лекарства трябва да се вземат предвид, въпреки че антибиотиците не трябва да се приемат заедно с алкохола. Ако имате пристъп, трябва да отидете в болницата или да се обадите директно на спешен лекар.
Освен това белите дробове трябва да бъдат пощадени, за да се избегне възпалението. Следователно, пациентите с дефицит на A-N-ацетилгалактозаминидаза не трябва да пушат при никакви обстоятелства. Здравословната диета и като цяло здравословния начин на живот имат много положителен ефект върху хода на заболяването.
Ако пациентът желае да има деца, генетичните консултации са полезни, за да се предотврати предаването на болестта на децата. Тъй като пациентите често страдат от ограничена подвижност, физиотерапията е полезна за облекчаване на това. Много упражнения могат да се изпълняват и в собствения ви дом.
Можете да направите това сами
Съществуващият дефицит на A-N-ацетилгалактозаминидаза е нелечимо заболяване. Следователно медицинското лечение е от съществено значение. Мерките за самолечение могат да се основават само на симптомите, за да се улесни ежедневието. Важно е родителите да наблюдават внимателно детето.
Родителите могат най-добре да помогнат на детето си чрез разбиране и търпение, любов и грижи. Освен това трудотерапията и логопедичните лечения винаги се основават на двойна концепция: терапия на място при специалиста и изпълнение на упражненията у дома. Родителите трябва да бъдат активни тук последователно и с търпение. Трябва да се осигури диета, богата на витамини и минерали, редовни упражнения и много чист въздух. Това укрепва имунната система и намалява риска от инфекция. Ако човекът има склонност към изземване, той не трябва да бъде сам дълго време и трябва да се провери дали те могат да се наранят, ако се появи гърч.
През повечето време пациентите не могат сами да се справят с ежедневието и се нуждаят от постоянна грижа. Ако родителите не могат да си позволят това - особено при по-големи деца и възрастни - те не трябва да се страхуват да потърсят помощ. Това може да бъде под формата на медицинска сестра или настаняване в подходящо заведение. Ако все пак искате да имате деца, се препоръчва консултация със специалист, тъй като заболяването се наследява.

.jpg)
.jpg)







.jpg)

.jpg)