Най- Мукополизахаридоза е събирателен термин за лизозомни заболявания за съхранение, които се основават на съхранението на гликозаминогликани. Всички заболявания развиват подобни симптоми и форми. Тежестта на синдромите варира значително.
Какво е мукополизахаридоза?

© Себастиан Каулицки - stock.adobe.com
А Мукополизахаридоза няма такова нещо като единична болест. Терминът мукополизахаридоза е събирателен термин за голям брой заболявания за съхранение, които се основават на нарушения на съхранението на гликозаминогликани (GAG) в лизозомите на клетките. Съхранението се осъществява постепенно, защото прекратяването на връзките не работи.
Всички мукополизахаридози са генетични. При всяка болест липсва специфичен ензим, който катализира разграждането на съответните GAG. Всички мукополизахаридози са много редки заболявания и често показват подобни курсове. Ако не се лекуват, непрекъснато нарастващите находища унищожават клетките. Органите се унищожават в процеса. Заболяването може да започне в ранна детска възраст, както и в зряла възраст.
Мукополизахаридозата може да бъде причинена от четири различни групи гликозаминогликани:
- Хепаран сулфат
- Кератан сулфат
- Хондроитин сулфат
- Дерматан сулфат.
Всички гликозаминогликани се състоят от полизахаридна верига, която е свързана с протеин. Въглехидратният компонент съставлява 95 процента, а протеиновият компонент - пет процента от молекулната маса. В зависимост от това кой гликозаминогликан и кой ензим е засегнат, мукополизахаридозите могат да бъдат разделени на шест различни основни форми: Те включват болест на Хърлер / Шеи (MPS I), болест на Хънтър (MPS II), болест на Санфилипо (MPS III), болест на Morquio ( MPS IV), болест на Maroteaux-Lamy (MPS VI) и болест на Sly (MPS VII). Всички видове имат тежки и леки форми.
каузи
Причината за всички мукополизахаридози е нарастващото съхранение на гликозаминогликани (GAGs) в лизозомите на клетките. Разграждането на съответните биополимери е нарушено. За всяко отделно разстройство липсва или определен ензим, или този ензим работи неправилно. Може да има няколко мутации за всеки ензим. Наследяването на съответната мутация може да бъде автозомно рецесивна, автозомно доминантна или х-свързана рецесивна.
Тъй като ензимният процес обикновено включва няколко етапа на реакция, няколко ензима могат теоретично да бъдат мутирани за един и същ гликозаминогликан. Симптомите на разстройството биха били еднакви или подобни.
- при MPS I, Болест на Хюрлер или Шейе, ензимът алфа-1-идуронидаза е дефектен.
- MPS II представлява синдромът на ловците с дефектна идуронат-2-сулфатаза.
- Синдромът на Санфилипо (MPS III) може да бъде разделен на няколко подтипа. При това състояние могат да бъдат засегнати няколко ензима.
- Болест на Morquio (MPS IV) се причинява от дефектна β-галактозидаза.
- При синдром на Марото-Лами (MPS VI) това е N-ацетил-галактозамин-4-сулфат сулфатаза.
- Болест на Слай (MPS VII) се причинява от дефектна β-глюкуронидаза. Когато съответните гликозаминогликани се съхраняват в лизозомите, те стават все по-големи и по-големи.
Клетките също се уголемяват, защото се нуждаят от повече и повече място за неразградени GAG. Това се забелязва и при разширяването на много органи. Типичен симптом е постоянното уголемяване на черния дроб и далака. Ако не се лекува, болестите за съхранение водят до смърт поради постепенното унищожаване на органите.
Симптоми, неразположения и признаци
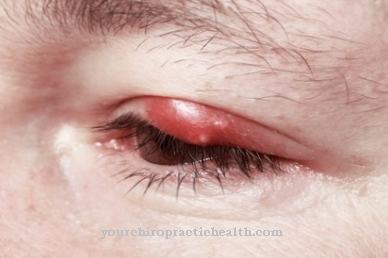
Симптомите са сходни за всички заболявания. Има тежки и леки форми. Лек курс обаче означава само, че болестта прогресира по-бавно. Крайният курс винаги е един и същ. Наблюдава се прогресивна деформация на скелетната система, ставни контрактури, груба черти на лицето и уголемяване на черния дроб и далака.
Умствените и двигателните умения намаляват в краткосрочен или дългосрочен план. При тежките форми на нарушенията клиничните картини са много сходни. Пъпните и ингвиналните хернии, сърдечните проблеми и респираторните инфекции се появяват на ранен етап. С течение на времето стесняването на дихателните пътища и разширяването на сливиците и сливиците се превръщат в масивни проблеми със сънна апнея.
Диагноза и ход на заболяването
Мукополизахаридозите могат да бъдат диагностицирани чрез изследване на урината за екскретирани гликозаминогликани. При мукополизахаридоза стойностите винаги се увеличават. Активността на предполагаемия дефективен ензим в левкоцитите или фибробластите също може да бъде определена. Определен модел на екскреция на гликозаминогликаните води до съмнението към съответния ензим, който след това се изследва.
Усложнения
В резултат на мукополизахаридоза засегнатите страдат от различни малформации и скелетни оплаквания. Появяват се деформации, които могат значително да ограничат ежедневието на пациента. По правило ставите са засегнати и от мукополизахаридоза, така че движението на пациента е ограничено.
По-специално децата са засегнати и страдат от силно забавено развитие, така че различни последващи увреждания могат да възникнат и в зряла възраст. Не са редки случаите, когато мукополизахаридозата може да причини проблеми в сърцето или дишането. В най-лошия случай внезапната сърдечна смърт може да доведе до смърт на съответния човек. Поради затрудненото дишане пациентите страдат от умора и умора.
Устойчивостта на засегнатите също намалява значително. Не са редки случаите, когато затрудненията с дишането през нощта водят до проблеми със съня и по този начин до депресия. Качеството на живот на пациента е значително намалено от мукополизахаридозата. Причинно-следственото лечение на това заболяване за съжаление не е възможно. Следователно засегнатите са зависими от донори на костен мозък за лечение на симптомите. Няма особени усложнения. Въпреки това, в повечето случаи пациентите са зависими от терапията през целия живот.
Кога трябва да отидете на лекар?
Промените и отклоненията в структурата на тялото показват нарушение на здравето. Посещението на лекар е необходимо веднага щом има постоянни оптични особености или засегнатото лице има проблеми с умишленото оптимизиране на стойката си.Подуване на ставите, промени в чертите на лицето или уголемяване на гърдите трябва интензивно да се изследват от лекар, за да може да се постави диагноза. Ако има ограничения в движението, нередности в ежедневния доброволен контрол и намаляване на физическите и умствените показатели, е необходим лекар. Трябва да се консултирате с лекар в случай на нарушения в сърдечния ритъм, проблеми с дишането или прекъсвания по време на нощен сън.
Подуване в гърлото, усещане за стягане в гърлото, нарушения в акта на преглъщане и промени във вокализацията се считат за тревожни. Те трябва да бъдат прегледани от лекар, за да могат симптомите да бъдат облекчени. Ако засегнатото лице претърпи повече инфекции, ако способността да се концентрира и обърне внимание намалява или ако пъпните или ингвиналните хернии се появяват многократно, лекарят трябва да бъде информиран за наблюденията.
Внезапни петна по кожата, пожълтяване на кожата, както и вътрешна неспокойност трябва да бъдат изследвани и лекувани. Необходим е лекар, веднага щом има болка в тялото, намалено качество на живот и проблеми в поведението. Ако има риск от задух, е необходима линейка. За да се предотврати това остро състояние, трябва да се консултирате с лекар възможно най-рано.
Терапия и лечение
Причинна терапия все още не е възможна. В изследователските проекти обаче има някои подходи за бъдеща генна терапия за тези заболявания. За съжаление, в момента няма осезаеми резултати в тази област. В Барселона обаче трябва да започне клинично проучване за генна терапия на болестта на Хюрлер. С някои форми на мукополизахаридози, трансферите в костния мозък се оказват ефективни в отделни случаи. Това се отнася например за болестта на Хънтър, болестта на Хърлер или болестта на Санфилипо.
Чрез този трансфер на костен мозък болните стволови клетки се обменят за здрави стволови клетки от донор. Това дава възможност на организма да възстанови достатъчно липсващия ензим. Ензимната заместителна терапия също се отплаща в много случаи. Тази заместителна терапия обаче трябва да се провежда за цял живот. Има обаче и случаи, в които обещаващи терапии вече не са възможни. Въпросът тук е да се провеждат симптоматични лечения.
Можете да намерите лекарствата си тук
➔ Лекарства за болкаПрогноза и прогноза
По-нататъшното развитие при пациенти с мукополизахаридоза трябва да се оценява индивидуално. Този термин е събирателен термин за различни заболявания при съхранение. Те присъстват в различна степен при всеки пациент и са индивидуално изразени по своята интензивност. Ако не се започне медицинска помощ, вътрешните органи на всички засегнати постепенно се разрушават в течение на живота си. Това води до съкращаване на средния очакван експлоатационен живот.
С ранна диагноза може да се разработи персонално оптимизирана терапия. Това е обвързано със здравните изисквания и съществуващите оплаквания на пациента. Дългосрочното лечение е от основно значение за постигане на стабилно подобряване на здравето. Могат да се появят хирургични интервенции, всяка от които е свързана с различни рискове и странични ефекти. Ако операцията протича без допълнителни усложнения, симптомите обикновено се облекчават след това.
Независимо от това, нежелани промени и пречки могат да възникнат в процеса на живот. В отделни случаи само трансплантацията на костен мозък може да подобри общото качество на живот. Поради общите обстоятелства пациентът изпитва голям емоционален и психически стрес. Нормалното ежедневие често не е възможно поради симптомите. Могат да възникнат психологически усложнения и да доведат до допълнително влошаване на ситуацията.
предотвратяване
Тъй като мукополизахаридозите са наследствени заболявания, профилактиката не е възможна. В случай на съществуващо заболяване, успехът на лечението може да бъде осигурен чрез навременна терапия. Освен това е необходимо постоянно наблюдение на белодробната и сърдечната функция. Ако в семейството вече са възникнали случаи на мукополизахаридоза, рискът от заболяването може да бъде оценен чрез генетична консултация, ако семейството желае да има деца.
Aftercare
В повечето случаи на мукополизахаридоза пациентът има само няколко възможности за последващи грижи, така че засегнатото лице трябва да се консултира преди всичко с лекар на ранен етап. Само с ранно откриване и лечение на това заболяване могат да бъдат предотвратени по-нататъшни усложнения, така че веднага след появата на първите признаци и симптоми трябва да се свърже с лекар.
В повечето случаи засегнатите са зависими от хирургични интервенции, които могат да облекчат и ограничат симптомите. Въпреки това, тъй като мукополизахаридозата е генетично заболяване, тя обикновено не може да бъде напълно излекувана.
Ето защо заинтересованото лице трябва първо да се консултира с лекар, ако иска да има деца, за да се предотврати повторната поява на това заболяване при децата. Често е много важно да имате подкрепа на семейството по време на лечението. Това също може да предотврати депресията и други психологически разстройства. Мукополизахаридозата може да доведе до намалена продължителност на живота на засегнатото лице, при което по-нататъшният курс зависи много от времето на поставяне на диагнозата.
Можете да направите това сами
Възможностите за самопомощ при мукополизахаридоза са ограничени до облекчаване на симптомите и по този начин подобряване на качеството на живот. Групите за самопомощ се оказаха много полезни, тъй като обменът с други родители разкрива ценни съвети и често може да облекчи страховете и притесненията и да даде по-позитивен поглед към бъдещето.
Придружаването към физиотерапия, трудотерапия, логопедия и други форми на терапия, които често могат да се задълбочат в домашната среда, сега са неразделна част от живота.
За да направите живота възможно най-лесен за себе си и засегнатото дете, препоръчително е да направите възможно най-рано жизнената среда достъпна за инвалидите. С увеличаването на възрастта и теглото на детето, леглата с грижа с регулиране на височината се оказват голямо физическо облекчение за възпитателя. Устройствата за предупреждение за епилепсия и други технически помощни средства позволяват най-добрата възможна безопасност дори през нощта и облекчават родителите през нощта, така че да могат да спят по-спокойно.
Воденето на дневник на симптомите може да помогне на лекаря да разпознае нови симптоми и евентуално да коригира лечението на съществуващите симптоми, тъй като лекарствената терапия често не показва желания, а обратния ефект.
Тъй като болестта е много взискателна към роднините, те трябва да създадат малки пространства за себе си, за да презаредят батериите си. Това може да включва лекове, превантивна грижа или по-късно ваканция в хосписа.
.jpg)










.jpg)

.jpg)