Най- Синдром на Смит-Лемли-Опиц е синдром на когенитална малформация. Причината е една от общо 70 генни мутации на хромозома 11q13.4. Заболяването се наследява по автозомно-рецесивен начин и е изключително рядко заболяване с множество малформации в органите и нарушена биосинтеза на холестерола.
Какво представлява синдромът на Смит-Лемли-Опиц?

© jijomathai - stock.adobe.com
Най- Синдром на Смит-Лемли-Опиц попада в групата на автозомно-рецесивните наследствени малформационни синдроми. Мутацията на ген причинява метаболитно разстройство в биосинтезата на холестерола при заболяването. Синдромът е най-честото когенитално разстройство на биосинтезата на холестерола. Разпространението на болестта в Европа е между около 1: 60 000 и 1: 10 000.
По този начин заболяването може да бъде класифицирано като рядко заболяване, въпреки че е едно от най-често срещаните когенитални заболявания при биосинтезата на холестерола. Синдромът е по-рядък на континентите Азия и Африка. Заболяването е описано за първи път през 1964 г. Генетиците Д. У. Смит, Л. Лемли и Дж. Мариус Опиц документират комплекса от симптоми от научна гледна точка. Оттогава са регистрирани малко над 300 случая.
Момчетата са по-често засегнати от момичетата. Симптомите вероятно са по-изразени при момичетата и затова са по-трудни за диагностициране. Заболяването е вродено, но се развива прогресивно от раждането нататък и поради това се характеризира с различни форми. Синдромът се разделя на типове I и II в зависимост от симптомите.
каузи
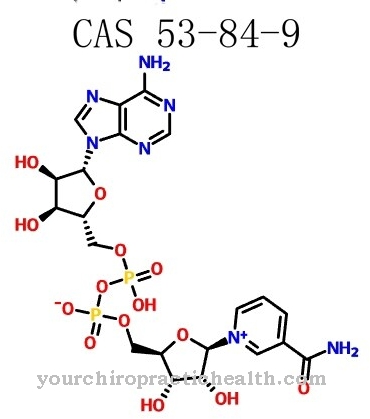
Синдромът на Смит-Лемли-Опиц се причинява от генна мутация, която е локализирана през 1998 г. Хромозома 11q13.4 сега се счита за местоположението на мутацията, като на това място досега са известни над 70 различни мутации. Видът на каузалната мутация определя тежестта и вида на симптомите във всеки отделен случай. Въпросният ген е 7-стерол редуктазен ген. S. Tint открива заедно с колегите си, че синдромът пречи на собственото производство на холестерол в организма.
Това производство включва превръщането на прекурсора 7-дехидрохостерол в собствен холестерол в организма, който не може да функционира поради ензимен дефект в резултат на мутацията. Следователно има излишък от 7-дехидрохостерол в организма на засегнатите. В същото време има общ дефицит на холестерола. Поради автозомното рецесивно наследяване на синдрома и двамата родители трябва да носят дефектния ген и могат да го предадат само на дете по този начин. Има 25 процента вероятност последващите деца на родители с болно дете също да бъдат засегнати от синдрома на малформация.
Симптоми, заболявания и признаци
Децата със синдром на Смит-Лемли-Опиц се раждат с типични краниофациални малформации, особено микроцефалия, изпъкнало чело и малък нос с широк носен корен. В допълнение към антеверситетните народи има микрогений. Често се наблюдават цепнато небце и замъгляване на лещата, особено катаракта и катаракта.
Има и блефароптоза. Психически и церебрални, нежелани развития се развиват с течение на времето, което води до умствен недъг. Холопронасефалията и раздразнителността също могат да оформят клиничната картина. Освен самонараняващото се поведение, синдромът може да провокира аутистично поведение. Освен това има множество малформации на органите, особено тези на сърцето и урогениталния тракт.
Хипоспадията и крипторхизмът са най-честите урогенитални малформации. В допълнение към излишните пръсти или пръсти на краката може да присъства синдактилия на пръстите на краката. Мускулната хипотония, смущения в преглъщането и гастроезофагеален рефлукс също са от значение в контекста на симптоматичния комплекс. Чревна дисмотилност и пилорна стеноза също са често срещани. При тип II на синдрома има псевдо-хермафродитизъм, при който външните гениталии са женски, въпреки че преобладава мъжкият каротип.
Диагноза и ход на заболяването
Като пренатална диагноза, ултразвуковите изследвания могат да запишат типичните физически характеристики на синдрома на Смит-Лемли-Опиц още преди раждането. Освен дефицит на растеж може да се забележи сърдечен дефект или липсата на бъбрек. При теста за амниотична течност мутационният анализ вече може да даде резултат, потвърждаващ диагнозата.
След раждането децата имат характерна форма на лицето и специални позиции на крайниците, така че подозираната диагноза може да бъде поставена чрез визуална диагноза, ако пренаталната диагноза не е била успешна. Генетичната диагноза обезпечава подозрението. По отношение на диференциалната диагноза, синдромът на Смит-Лемли-Опиц трябва да се разграничи от фетален алкохолен синдром, синдром на Палистър Хол, синдром на Кауфман-Маккусик и синдром на Корнелия де Ланге.
Синдромът на Pätau, синдромът ATR-X и синдром C, синдромът на Zellweger и синдром на хидролетхала също трябва да се вземат предвид при диференциалната диагноза. Същото се отнася и за синдрома на Oro-Faciales-Digitales, синдрома на Holoprosencephaly-Polydactyly и Meckel-Syndrome. Продължителността на живота на децата зависи от концентрацията на холестерола и лечимостта на малформациите на органите. Ниските нива на холестерол и най-тежките малформации правят скоро вероятния фатален изход. Децата с високи нива на холестерол и лесно лечими малформации нямат силно нарушена продължителност на живота.
Усложнения
Поради синдрома на Смит-Лемли-Опиц засегнатите страдат от различни малформации и деформации. Те имат много негативен ефект върху качеството на живот на пациента. По-специално вътрешните органи са засегнати от малформациите, така че смъртта често може да настъпи веднага след раждането. Освен това повечето пациенти страдат от цепка на небцето, а също и от проблеми с очите.
Освен това този синдром често води до интелектуални затруднения и по този начин до интелектуална изостаналост. Поради това повечето пациенти са зависими от помощта на други хора в живота си и вече не могат да се справят сами с много ежедневни задачи. Сърцето също е засегнато от малформациите, които могат да доведат до внезапна сърдечна смърт. Освен това синдромът на Смит-Лемли-Опиц засяга и гениталиите, така че при тях могат да се появят и малформации.
Лечението на синдрома на Смит-Лемли-Опиц обикновено може да бъде само симптоматично. Няма усложнения и някои от симптомите могат да бъдат ограничени. Въпреки това не може да се постигне напълно положителен ход на заболяването. Не може да се предвиди универсално дали продължителността на живота ще бъде ограничена.
Лечение и терапия
За пациенти със синдром на Смит-Лемли-Опиц, социалните и медицински грижи през целия живот често са неизбежни. По правило развитието им се забавя силно по отношение на познавателната и двигателната област. Почти във всички случаи това води до увреждане през целия живот, което не позволява самостоятелен начин на живот. Затова преди всичко се осигурява поддържащо лечение.
Като част от тези мерки родителите получават психотерапевтична подкрепа и в идеалния случай се учат да се справят със заболяването на детето си. Синдромът на Смит-Лемли-Опиц е нелечим и затова не може да се третира като причина. Тъй като за синдрома е документирано нарушение на метаболизма на холестерола, е възможно симптоматично лечение за компенсиране на дефицита на холестерол. Това лечение се извършва чрез даване на холестерол.
Най-многобройните малформации на органите трябва да бъдат коригирани хирургически, доколкото това е възможно. Изключение от това е често документираното многовързано свързване на пръстите и пръстите на краката, което не изисква непременно хирургическа намеса. Съпътстващите симптоми като зрителни затруднения сега също могат да бъдат лекувани и облекчени добре.
По-голямата част от засегнатите страдат от хранителни проблеми като затруднения в смученето и преглъщането или гастроезофагеален рефлукс и нарушена стомашно-чревна перисталтика. Затова често трябва да се използва стомашна тръба, за да се гарантира безопасен прием на храна. Проблемите в поведението е възможно да се лекуват с поведенческа терапия.
предотвратяване
След положителна пренатална диагноза за синдрома на Смит-Лемли-Опиц, родителите получават възможност да прекратят бременността. Синдромът на Смит-Лемли-Опиц може да бъде предотвратен по някакъв друг начин, само ако двойките имат молекулярно-генетична диагноза, извършена в семейното им планиране и след като бъдат представени доказателства за мутацията, вземат решение срещу собствените си деца.
Aftercare
Последващите мерки за синдрома на Смит-Лемли-Опиц (SLOS) се основават на тежестта на симптомите, възникващи в хода на заболяването. В по-голямата част от случаите на заболяване децата имат хранителни проблеми. Те се справят зле. Следователно фокусът на последващите грижи е на първо място адекватното хранене на засегнатите деца с помощта на назогастрални висококалорични течни храни и прилагане на достатъчно холестерол.
По-нататъшното протичане на заболяването показва и при много от засегнатите деца недоразвитие на мозъка. Това недоразвитие обикновено води до физически или психически увреждания с различна степен на тежест. Например, не всички засегнати деца се учат да ходят. За компенсиране на ограничената мобилност в този случай се предвиждат помощни средства за ежедневно движение (напр. Инвалидна количка, помощни средства за ходене и стояне).
В случай на психични симптоми като автоагресия и хиперактивност, терапевтично предписаното лечение с лекарства продължава като последваща мярка. Освен това около 50 процента от всички засегнати деца ще развият умерен до тежък сърдечен дефект с напредването на болестта. След операцията на сърдечния дефект се правят редовни електрокардиографски изследвания (ЕКГ) и сонографски изследвания.
За родители на деца със SLOS се препоръчват психологически консултации и терапии. Независимото живеене в зряла възраст е малко вероятно. Трябва да се очакват обширни мерки за грижа при последващи грижи за SLOS в зряла възраст. В допълнение, малформациите на органите могат да ограничат продължителността на живота.
Можете да направите това сами
Заболяването е свързано с многобройни оплаквания, които силно влошават качеството на живот. Ако член на семейството е диагностициран с генетично заболяване, преди зачеването на детето трябва да се консултира с лекар. Възможните рискове трябва да бъдат претеглени, за да могат да се вземат разумни решения за всички участващи. В допълнение, трябва да участвате във всички проверки, предлагани по време на бременност.
Веднага щом се забележат нарушенията на здравето на детето, родителите и близките могат да предприемат подходящи предпазни мерки и да се подготвят по-добре за бъдещото развитие. В голям брой случаи грижата за някой със синдрома на Смит-Лемли-Опиц е огромно предизвикателство за близките. Затова те трябва да знаят и да се придържат към своите физически и емоционални граници. Препоръчително е да се обмисли използването на медицинска помощ за пациента и паралелно с психотерапевтичната подкрепа за близките. В резултат на това често могат да бъдат постигнати подобрения при справяне с болестта.
Трябва да се отбележи, че стабилността на социалната среда е важна за пациента. Освен това винаги трябва да запазите спокойствие в ежедневието, когато възникнат неприятности и предизвикателства. Тъй като засегнатото лице не е в състояние да оформи живота си независимо, трябва да има специална съпричастност към отношенията с него.
.jpg)









.jpg)

.jpg)